Zooming into Specific Regions & Comprehensive Visualisation
This example focuses on zooming.
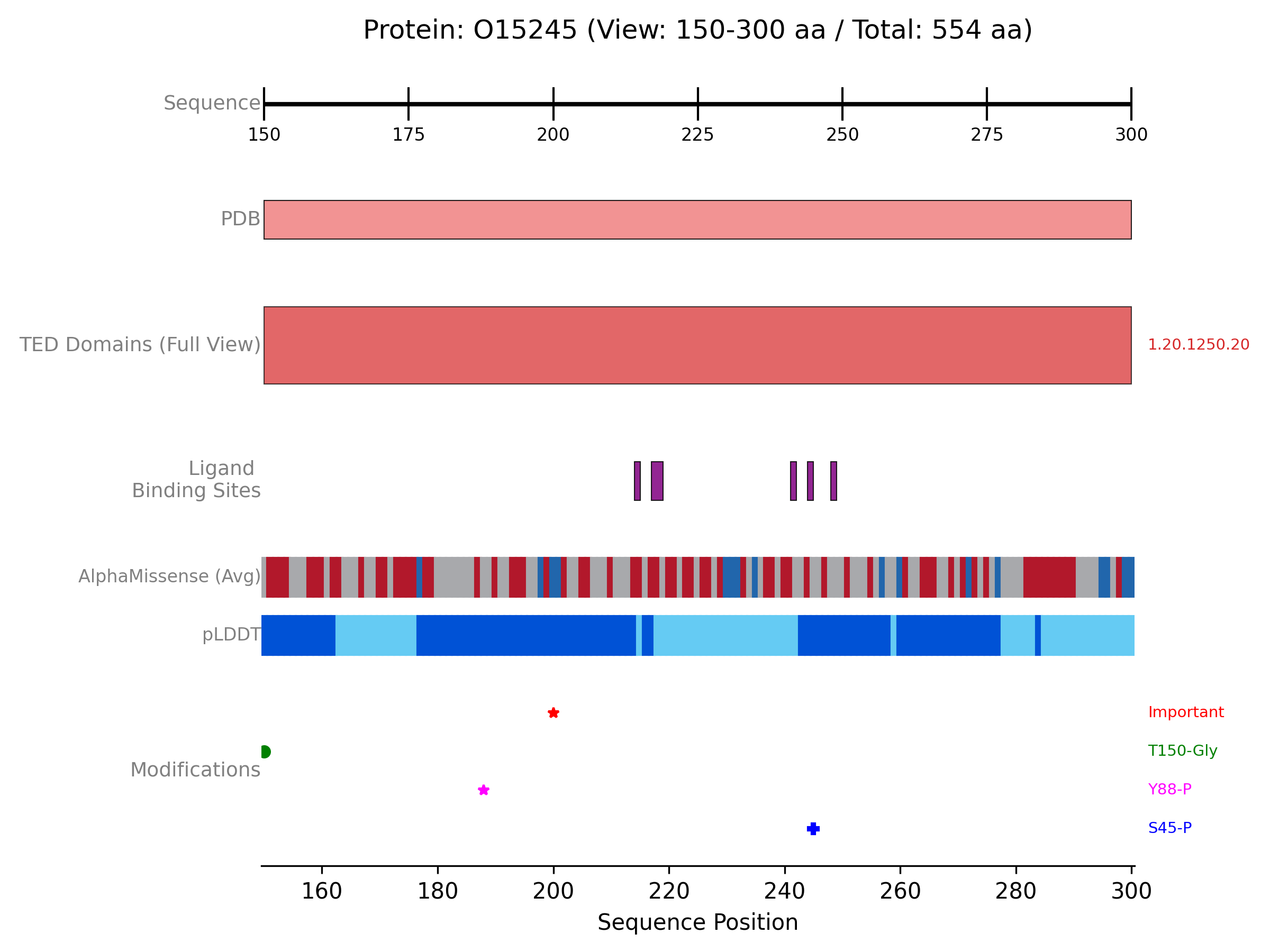
Protviz allows you to focus on specific areas of a protein sequence for a more detailed inspection of annotations. This is achieved by providing view_start_aa and view_end_aa arguments to the plot_protein_tracks function.
The following example demonstrates how to create a comprehensive plot for the protein “O15245”, fetching data from PDBe, AlphaFold Database, and the TED database. It then generates a zoomed-in view of a specific region (residues 150-300, adjusted if the protein is shorter), displaying PDB coverage, TED domains, ligand interactions, AlphaFold metrics (pLDDT and AlphaMissense), and custom PTM annotations.
Example: Multi-Track Plot with Zoom
from protviz import plot_protein_tracks
from protviz.data_retrieval import AFDBClient, PDBeClient, get_protein_sequence_length, TEDClient
from protviz.tracks import (
AlphaFoldTrack,
AxisTrack,
CustomTrack,
LigandInteractionTrack,
PDBTrack,
TEDDomainsTrack,
)
def main():
uniprot_id = "O15245" # Target protein UniProt ID
# 1. Initialise Data Retrieval Clients
pdbe_client = PDBeClient()
afdb_client = AFDBClient()
ted_client = TEDClient()
try:
print(f"--- Protein: {uniprot_id} ---")
# 2. Fetch Overall Protein Information
seq_length = get_protein_sequence_length(uniprot_id)
print(f"Sequence length for {uniprot_id}: {seq_length}")
# 3. Fetch Data from Various Sources
print(f"\nFetching PDB ligand interactions for {uniprot_id}...")
ligand_data = pdbe_client.get_pdb_ligand_interactions(uniprot_id)
if ligand_data:
print(f"Found {len(ligand_data)} ligand interaction contexts.")
else:
print(f"No ligand interaction data found for {uniprot_id}.")
print(f"\nFetching PDB coverage for {uniprot_id}...")
pdb_coverage_data = pdbe_client.get_pdb_coverage(uniprot_id)
if pdb_coverage_data:
print(f"Found {len(pdb_coverage_data)} PDB entries.")
else:
print("No PDB coverage data found.")
print(f"\nFetching AlphaFold data for {uniprot_id} (pLDDT and AlphaMissense)...")
alphafold_data = afdb_client.get_alphafold_data(
uniprot_id, requested_data_types=["plddt", "alphamissense"]
)
# (Optional: print statements for AlphaFold data counts)
print(f"\nFetching TED domain annotations for {uniprot_id}...")
ted_domain_annotations = ted_client.get_TED_annotations(uniprot_id)
if ted_domain_annotations:
print(f"Found {len(ted_domain_annotations)} TED domain annotations.")
else:
print(f"No TED annotation data found for {uniprot_id}.")
# 4. Define Zoom Region
# Initial desired zoom start and end amino acid positions
zoom_s, zoom_e = 150, 300
# Adjust zoom window if the protein is shorter than the desired end
if seq_length < zoom_e:
zoom_s = seq_length // 4 # Start at one quarter of the sequence length
zoom_e = (seq_length // 4) * 3 # End at three quarters
# Ensure start is less than end, and handle very short sequences
if zoom_s >= zoom_e:
zoom_s = 1
zoom_e = seq_length
print(f"\nPlotting Zoomed View for region: {zoom_s}-{zoom_e}")
# 5. Create Tracks for the Visualisation
# Axis track, always showing the context of the total sequence length
axis_trk_zoom = AxisTrack(
sequence_length=seq_length, # Total length for correct scaling context
label="Sequence",
)
# PDB coverage track, collapsed for a summary view within the zoom
pdb_trk_detail_zoom = PDBTrack(
pdb_data=pdb_coverage_data,
label="PDB",
plotting_option="collapse",
color="lightcoral",
)
# TED domains track, shown in "full" detail
ted_domains_trk_detailed = TEDDomainsTrack(
ted_annotations=ted_domain_annotations,
label="TED Domains", # Adjusted label for clarity
plotting_option="full",
show_domain_labels=True,
)
# Ligand interaction track, collapsed for the zoomed view
ligand_trk = LigandInteractionTrack(
interaction_data=ligand_data,
label="Ligand Binding", # Shortened label for space
show_ligand_labels=True, # Still useful if space permits in "full"
plotting_option="collapse", # Changed to collapse for zoomed summary
site_height=0.1,
)
# AlphaFold track showing both pLDDT and AlphaMissense
alphafold_track = AlphaFoldTrack(
afdb_data=alphafold_data,
plotting_options=["plddt", "alphamissense"],
main_label="", # Minimal label if space is tight or redundant
plddt_label="pLDDT",
alphamissense_label="AlphaMissense", # Simplified label
sub_track_height=0.1,
sub_track_spacing=0.05
)
# Custom track for Post-Translational Modifications (PTMs)
ptm_annotations = [
{"position": 245, "label": "S245-P", "color": "blue", "display_type": "marker", "marker_symbol": "P"},
{"position": 188, "label": "Y188-P", "color": "#FF00FF", "display_type": "marker", "marker_symbol": "*"},
{"position": 150, "label": "T150-Gly", "color": "green", "display_type": "marker", "marker_symbol": "s", "marker_size": 5}, # Example of a different marker
{"position": 200, "label": "Important", "color": "red", "display_type": "marker", "marker_symbol": "X", "marker_size": 7},
]
ptms_track = CustomTrack(
annotation_data=ptm_annotations,
label="Modifications",
ann_height=0.05,
show_row_labels=False, # Assuming PTMs don't need row labels here
padding=0.05,
)
# 6. Plot the Tracks with Zoom Parameters
print("\nPlotting...")
plot_protein_tracks(
protein_id=f"{uniprot_id}_zoomed", # Indicate zoom in filename
sequence_length=seq_length,
tracks=[ # Order of tracks matters for display
axis_trk_zoom,
pdb_trk_detail_zoom,
ted_domains_trk_detailed,
ligand_trk,
alphafold_track,
ptms_track,
],
view_start_aa=zoom_s, # Start of the zoomed region
view_end_aa=zoom_e, # End of the zoomed region
figure_width=10, # Adjusted figure width for zoom
# figure_height=6, # Explicitly set figure height if desired
save_option=True
)
print(f"Zoomed plot saved as {uniprot_id}_zoomed_plot.png")
except Exception as e:
print(f"An error occurred: {e}")
import traceback
traceback.print_exc()
if __name__ == "__main__":
main()
The previous example will generate a plot like this one: